Simulationen im Bereich wo die chemische Struktur der Moleküle schon erkennbar ist, aber das System als solches noch weitgehend klassisch beschrieben werden kann, umfassen in der Regel einige hundert bis tausend Moleküle unter möglichst realistischen Bedingungen. Solche realistischen Bedingungen lassen sich mit sogenannten Molekulardynamik (MD) oder Monte Carlo (MC) Algorithmen realisieren (vgl. oben). Die mikroskopischen bzw. makroskopischen Eigenschaften solch einer geringen Substanzmenge lassen sich mit Methoden der Statistischen Physik analysieren und anschließend mit tatsächlichen Experimenten vergleichen.
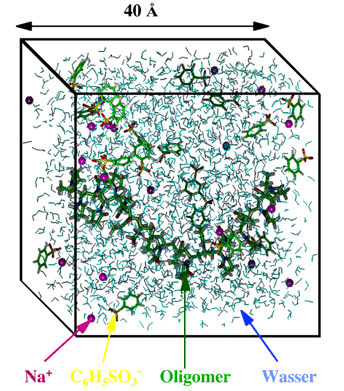
Betrachten wir dazu einige Beispiele. Die folgende Abbildung zeigt ein würfelförmiges Simulationsvolumen gefüllt mit Wasser (dargestellt durch die blauen Dreiecke). In dem Wasser sind Moleküle gelöst. Durch die Verwendung von periodischen Randbedingungen ist das System quasi unendlich ausgedehnt.
Abbildung 1: Ein Beispiel für ein würfelförmiges Simualtionsvolumen gefüllt mit Wasser und anderen Molekülen:
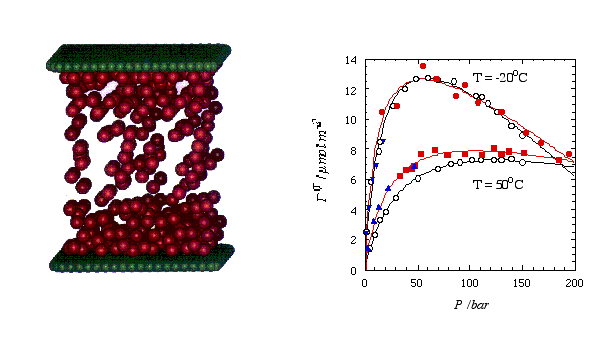
Abbildung 2 zeigt eine Momentaufnahme aus einer Molekulardynamik-Simulation von Methanmolekülen, die sich zwischen zwei Graphitoberflächen befinden. In der Simulation werden periodische Randbedingungen parallel zu den Oberflächen verwendet, so dass das simulierte System einem quasi unendlich ausgedehnten Schlitz entspricht. Man erken-nt die Anhäufung von Methanmolekülen an den Oberflächen. Dieser Konzentrationsunterschied im Vergleich zum Zentrum des Schlitzes nennt man den Oberflächenüberschuß. Ebenfalls in Abbildung 3 ist dieser als Funktion des Bulk-Drucks P gezeigt. Experimentell ist P der Methandruck weit entfernt von der Oberfläche. In der Simulation wird er in der Mitte zwischen den beiden Oberflächen bestimmt, wo die Bedingungen ausreichend genau dieser experimentellen Situation entsprechen. Die so gewonnenen simulierten Adsorptionsisothermen stimmen gut mit den experimentellen Messungen überein. In einem sochen Fall ist die Simulation schneller und einfacher durchzuführen als das Experiment.
Abbildung 2 Links: Momentaufnahme einer Simulation von Methanmolekülen (rote Kugeln) zwischen zwei Graphitoberflächen (durch die grünen Kugeln repräsentiert). Rechts: Oberflächenüberschuß als Funktion des Drucks von Bulk-Methan für zwei Temperaturen. Die offenen Kreise (angepasst mit den schwarzen Linien) sind experimentelle Resultate. Die geschlossenen Symbole (angepasst mit den roten Linien) stellen die entsprechenden Simulationsergebnisse dar:

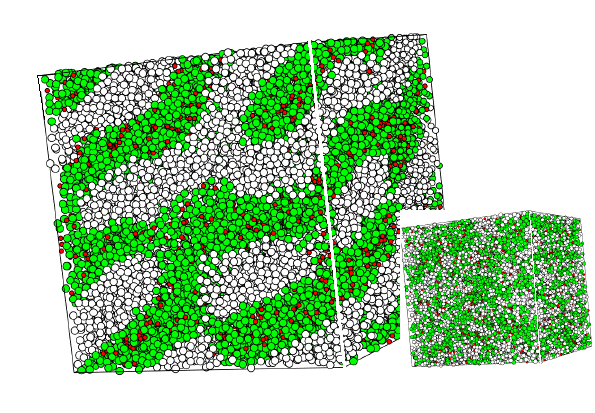
Abbildung 3 zeigt das Simulationsergebnis für die aus Lösung adsorbierte Monolage eines komplizierteren Systems, ein Cyclodextrinderivat auf Gold (vgl. kleines Bild; Substrat ist nicht dargestellt). Die Simulation sagt hier eine Doppelschichtstruktur der Monolage voraus, die sowohl die experimentelle Belegungsdichte als auch die Schichtdicke sehr gut reproduzieren kann. Diese Art Simulationen sind nützlich, wenn es um das Verständnis der Wechselwirkung von Rezeptorschichten mit Gastmolekülen geht.
Simulation eines Cyclodextrinderivates (kleines Bild) auf Gold. Oben: separate Darstellung der beiden Lagen innerhalb der Monoschicht. Unten: Die komplette Monoschicht.
Das folgende Beispiel illustriert eine Anwendung des Molekularen Modellierens auf das Phasenverhalten eines einfachen Modelltensids. Tenside sind sogenannte amphiphile Moleküle, die sich aus einem wasserabstoßenden und einen "wasserliebenden" Teil aufbauen. Abbildung 4 zeigt das Resultat einer MD-Simulation für ein System aus ca. 30000 Zentren bei einem Tensid-Molenbruch von 0.5. Durch die drastische Vereinfachung der Tensidmoleküle, der wasserabstoßende und der "wasserliebende" Teil jedes Tensidmoleküls wird jeweils mit vier Zentren dargestellt, kann ein recht großes System modelliert werden. Wie die Abbildung zeigt, bilden die Tensidmoleküle unter den gewählten Bedingungen ein lamellares Netzwerk (man spricht auch von Mikrophasenentmischung). Die Wassermoleküle (jeweils ein rotes Zentrum) halten sich zwischen den wasserliebenden Tensidzentren auf. In Abhängigkeit vom Molenbruch, von der Temperatur oder aber von der Architektur des Modelltensids können sich auch isolierte Aggregate oder andere Nanostrukturen bilden. Ziel solcher Simulationen ist es, ein Phasen-diagramm des realen Systems aufzustellen.
Abbildung 4 MD-Simulation eines Systems aus 30000 Zentren entsprechend 3350 Tensid- und 3200 Wassermolekülen. Das Bild zeigt lamellare Strukturen in der Nähe des Gleichgewichts. Der Einsatz unten rechts zeigt die zufällig verteilte Anfangskonfiguration.

Abbildung 4a MD-Simulation wie in Abbildung 4 aber mit mehr Wassermolekülen (rot).
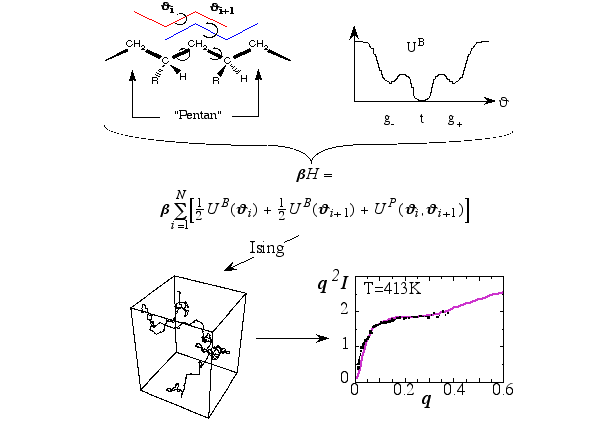
Die letzte Abbildung illustriert Schritte in der Modellbeschreibung eines Kunststoff- oder besser Polymermoleküls in der Schmelze. Oben links ist die schematische Darstellung eines linearen Makromoleküls gezeigt, dessen Konformationen durch die Torsionswinkel entlang seiner Hauptkette bestimmt sind. Ein Beispiel für eine Konformation ist in dem Würfel unten links gezeigt. Sind die innermolekularen Kräfte bzw. Potentiale bekannt (ein einfaches Torsionspotential ist oben rechts gezeigt), dann läßt sich die Berechnung der Polymerkonformationen auf das sogenannte Ising-Modell aus der Statistischen Mechanik abbilden. Mithilfe der so erzeugten Polymerkonformationen kann deren Röntgen- oder Neutronenstreuung berechnet und mit dem Experiment verglichen werden. Der Graph unten rechts zeigt die Streuintensität I multipliziert mit dem Betragsquadrat des Streuvektors q aufgetragen gegen q. Die durchgezogene Linie ist die theoretische Kurve. Die Punkte sind experimentelle Werte. Derartige Untersuchungen dienen dem Verständnis vom Zusammenhang zwischen makromolekularer Struktur und den Eigenschaften von Polymermaterialien.
Abbildung 5 Illustration der Schritte in der Modellbeschreibung eines Kunststoff- bzw. Polymermoleküls.